

NANOSCALE THEORY, MODELLING AND COMPUTATION
The research activities in the thematic area of nanoscale theory modelling and
computation are aimed at developing theoretical and computational methodologies as well as
their implementation in the related high performance computing software, to model and
predict phenomena and experiments of systems at the nanoscale. These include
first-principles approaches for molecules, small nanoparticles and materials; atomistic and
coarse-grained molecular dynamics simulations for (bio)molecules; density functional theory
approaches to study electronic, optical and magnetic properties of nanosystems and
molecules; novel theoretical approaches to simulate the real time evolution of molecules
interacting with plasmons and light; effective-mass schemes to handle complex nanostructures
that are beyond the reach of first-principles tools. The impact of these methodologies ranges
across various applications spanning from medicine to energy conversion, quantum optics and
telecommunication, optical sensing and molecular spintronics.
1. Developments of advanced DFT methodologies.
Contact person:
Arrigo Calzolari
Stefano Corni
DFT has been extended to include the description of additional quantum states by introducing advanced functional forms. Recent theoretical developments have demonstrated that starting from
standard results of first-principles simulations, it is possible to derive two estimators, namely
aplasmonicity index and a “natural” metric distance of electronic correlations, to quantify the
plasmonic character of optical excitations in nanostructures and the internal correlationsin
different materials, respectively.
Plasmonicity Index in Ag and Si nanoclusters:
|
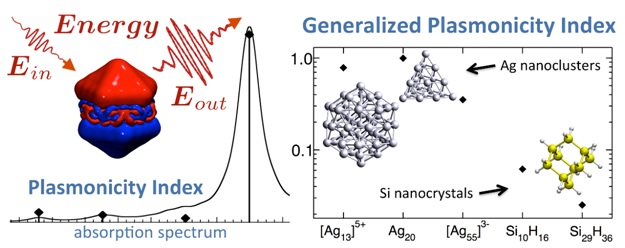
|
|
TDDFT absorption spectrum (black line) and PI
analysis (black diamonds and vertical lines) of Ag20.
Comparison of GPI values computed for selected excitations of Ag and Si nanoclusters. The
atomic structures of some clusters are shown as inset.
L. Bursi, A. Calzolari, S. Corni,
and E. Molinari, Quantifying the
Plasmonic Character of Optical Excitations in Nanostructures, ACS Photonics , 3,
520-525 (2016)
R. Zhang, L. Bursi, J. D. Cox, Y. Cui, C. M. Krauter, A. Alabastri, A. Manjavacas, A.
Calzolari, S. Corni, E. Molinari, E. A. Carter, F. J. García de Abajo, H. Zhang, and P.
Nordlander, How To Identify
Plasmons from the Optical Response of Nanostructures, ACS Nano, 11, 7321-7335
(2017)
|
|
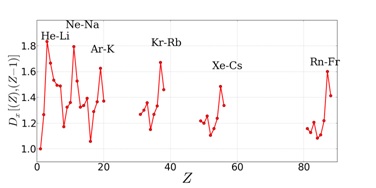
|
|
The distance, Dx, is computed between atoms
with atomic numbers Z and Z-1 and is plotted against Z for the s and p blocks of the periodic
table. The curve peaks when considering the last atom of one row and the first of the next.
The expected periodicity is well reflected in the behaviors of Dx.
S. Marocchi, S.
Pittalis, and I. D'Amico Fermionic correlations as metric
distances: A useful tool for materials science, Phys. Rev. Materials, 1, 043801
(2017)
|
2. Electronic, Magnetic, and Optical Properties.
Contact person:
Marco Affonte
Valerio Bellini
Deborah Prezzi
Novel ab-initio modeling for the optical time resolved experiments applied to low dimensional systems, have revealed the importance of many body effect even in the low pumping regime. Ab initio
ground and excited-state calculations are able to clarify the role of quantum confinement
effect and of the surface orientation, in anatase nanosheets. Large scale DFT simulations have
captured the microscopic mechanisms behind the magnetic coupling between magnetic
molecules and substrate, and have suggested possible switching mechanisms, a key element in
the realization of functional molecular magnetic devices.
|
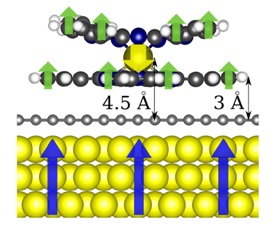
|
|
Adsorption geometry of the TbPc2 on graphene/Ni(111). The arrows represent the three different spins: the Tb, the radical on the Pcs and the Ni slab. The orientation of the spins is the one attained in the ground state, as calculated by DFT.
S. Marocchi, A. Candini, D. Klar, W. Van den Heuvel, H. Huang, F. Troiani, V. Corradini, R. Biagi, V. De Renzi, S. Klyatskaya, K. Hummer, N. B. Brookes, M. Ruben, H. Wende, U. del Pennino, A. Soncini, M. Affronte and V. Bellini, Relay-like exchange mechanism through a spin radical between TbPc2 molecules and graphene/Ni(111) substrates, ACS Nano, 10, 9353 (2016)
|
|
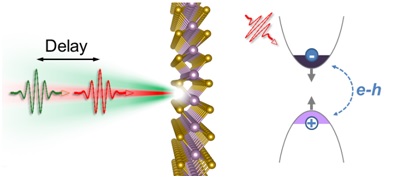
|
|
Ab initio modeling of the ultrafast optical response in single layer MoS2 is able to capture the main feature observed in pump-probe experiments, by accurately describing the bandgap renormalization due to photoexcited carriers.
E.A.A. Pogna, M. Marsili, D. De Fazio, S. Dal Conte, C. Manzoni, D. Sangalli, D. Yoon, A. Lombardo, A. C. Ferrari, A. Marini, G. Cerullo, and D. Prezzi Photo-Induced Bandgap Renormalization Governs the Ultrafast Response of Single-Layer MoS2, ACS Nano, 10, 1182
(2016)
|
|
|
|
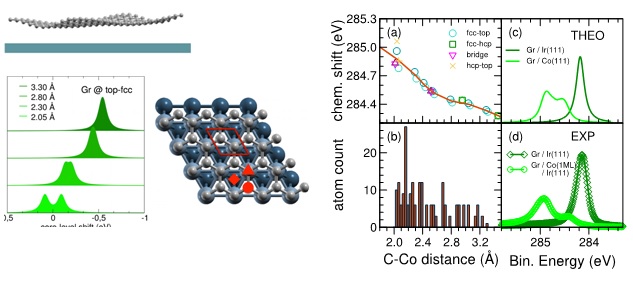
Top left panel: a cartoon of the corrugated graphene as given by the egg-box model. Bottom left panels: C1s core-level shifts (CLS) computed for the two non-equivalent C atoms of Gr@Co(0001) in top-fcc registry (C atoms at fcc-hollow and on-top sites), with increasing graphene-Co distance and represented as Lorentzian functions with a width of 0.1 eV. The average of the CLS computed for GR@top-fcc at 2.05 Ang distance was used as reference, here set to zero. Aside: top view of the studied fcc-top Gr@Co(0001) geometry with grey representing C and blue Co atoms. The top Co layer is represented in lighter blue. The 1x1 unit cell is reported. Circle and triangle symbols refer to fcc and top adsorption sites, respectively. Diamond symbols correspond to the hcp site.
Right panels: (a) Core-hole shift dependence with C-Co distance computed for Gr@Co. The red line gives the average of the values computed for the two non-equivalent C sites of four different registries of commensurate Gr@Co, shown by the symbols in different colours. (b) Height distribution for corrugated Gr@Co as given by the egg-box model. (c) XPS spectra computed as a sum of Lorentzian functions centred in the core-hole energies given in the left panel. (d) Experimental C1s XPS spectra.
G. Avvisati, S. Lisi, P. Gargiani, A. Della Pia, O. De Luca, D. Pacile, C. Cardoso, D. Varsano, D. Prezzi, A. Ferretti, and M.G. Betti, FePc adsorption on the moiré superstructure of graphene intercalated with a Co layer, J. Phys. Chem. C , 121, 1639
(2017)
|
3. Effective-mass schemes, Model Hamiltonians, and Many-Body Physics in
Nanosystems.
Contact person:
Massimo Rontani
Elisa Molinari
Powerful non ab-initio approaches are applied to complex
nanosystems such as quantum wires, dots, carbon nanotubes, two-dimensional structures,
which focus on the relevant low-energy scales to highlight complex collective quantum
behavior and novel many-body insulating phases.
|
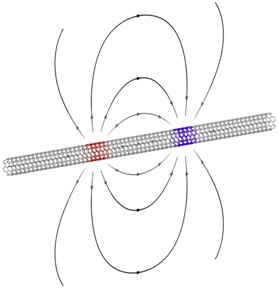
Sketch of a suspended armchair carbon nanotube.
The field lines of the Coulomb force between electron and hole lie mainly in the vacuum, hence screening is heavily suppressed
|
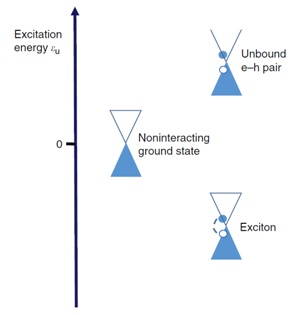
Excitonic instability of an armchair
carbon nanotube
|
D. Varsano, S. Sorella, D. Sangalli, M. Barborini, S. Corni, E. Molinari, and M. Rontani,
Carbon nanotubes as excitonic insulators, Nat.Commun, 8, 1461 (2017)
4. Coarse-Grained Force Fields.
Contact person:
Riccardo Nifosì
Valentina Tozzini
Giorgia Brancolini
Computational modeling of the membrane penetration mechanisms by peptide-aggregate could be greatly facilitated by using simplified coarse grain (CG) models. Recent strategy have been developed to build and optimize statistics based analytical CG force fields, particularly suited to account for the common interaction motives between biopolymers.
|
|
|
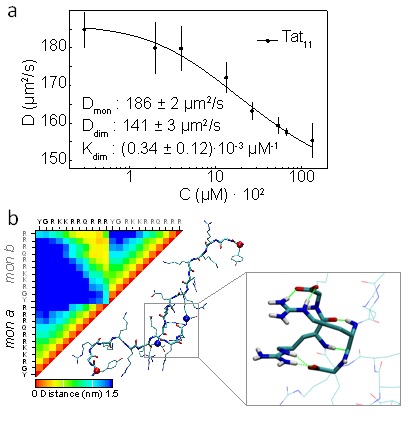
(a) Diffusion coefficients obtained by NMR DOSY experiments for increasing peptide concentrations. Monomer and dimer diffusion coefficients with dimerization constant are obtained by fitting.
(b) Tat11 inter and intra peptide contact map averaged during the simulation, and dimer representative structure. Single letter amino acids are indicated for each monomer. The inset shows the Arg-Arg stacking motif with salt bridges between C-termini and Arg side chains.
S. Macchi, R. Nifosi, G. Signore, S. Di Pietro, C. Boccardi, F. D'Autilia, F. Beltram, and F. Cardarelli, Self-aggregation propensity of the Tat peptide revealed by UV-Vis, NMR and MD analyses, Phys. Chem. Chem. Phys., 19, 23910 (2017)
|
|
|
|
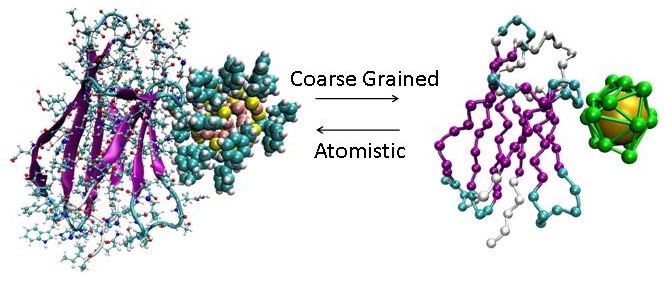
Development of a specific Coarse Grained approach to achieve an accurate description of protein-surface systems. The novelty of the research proposal respect to the state-of-the-art, is the description and parameterization of protein-NP interaction based on data obtained from docking and enhanced sampling molecular dynamics including information about the diffusivity of the gold NPs and protein in solution.
V. Tozzini, G. Brancolini, Multi-Scale Modeling of Proteins interaction with Functionalized Nanoparticles, Current Opinion in Colloid & Interface Science (2018).
|
|